

搜索网站、位置和人员



新闻活动开云体育软件官网
近日,西湖大学理学院、人工光合作用与太阳能燃料中心孙立成团队在《Journal of the American Chemical Society》上发表了题为“Cluster-Level Heterostructure of PMo12/Cu for Efficient and Selective Electrocatalytic Hydrogenation of High-Concentration 5‑Hydroxymethylfurfural”的研究论文。
研究团队通过在Cu纳米线表面负载Keggin型磷钼酸(H3PMo12O40,PMo12)团簇,制备了团簇级异质结构的PMo12/Cu催化剂。该催化剂能够将5-羟甲基糠醛(HMF)电催化加氢至2,5-二羟甲基呋喃(BHMF),并且在高底物浓度(1.0 M)下表现出前所未有的高选择性,法拉第效率高达98%,BHMF的生成速率高达4.35 mmol cm–2 h–1。相关工作不仅为醛类化合物电催化选择性加氢催化剂设计提供了新思路,还为电催化剂异质界面工程调控催化活性与选择性带来了更深的认识。
西湖大学博士后曹兴为文章第一作者,中国科学院院士、西湖大学理学院化学讲席教授、西湖大学人工光合作用与太阳能燃料中心主任孙立成为文章通讯作者。

加氢反应是合成众多化学品的重要工业反应。例如,从生物质中衍生的平台化合物5-羟甲基糠醛(HMF)可以通过选择性加氢转化为更高价值的产品2,5-二羟甲基糠醛(BHMF)。这种可再生二醇化合物可以代替化石来源的二醇单体,应用于功能化聚酯、聚氨酯以及药物中间体的合成。电化学加氢为实现5-羟甲基糠醛为代表的醛类化合物加氢至高附加值醇类提供了一条可持续的途径。然而,在高浓度下极易发生的偶联副反应,严重阻碍了其实际应用。
西湖大学人工光合作用与太阳能燃料中心孙立成院士团队通过分析HMF电化学加氢反应机制发现,HMF中的醛基电化学加氢生成醇涉及两步活性氢原子转移(HAT)过程加氢反应,当催化剂不能提供足够的H*时,第二步加氢反应受到阻碍,导致第一步加氢生成的烷基中间体偶联至双(羟甲基)呋喃。HAT过程通常遵循Langmuir-Hinshelwood(L-H)机制,高底物浓度下,水分子和HMF之间存在吸附竞争,会进一步降低H*的生成。基于此,孙立成院士团队提出针对催化剂的设计,不仅需要提高H*在催化剂表面的覆盖度,更加关键的是如何降低烷基中间体加氢反应能垒,减少其在催化剂表面的累积。针对这一重要的科学问题,该团队通过构造PMo12/Cu团簇级异质界面为加氢反应提供全新的反应位点,界面处独特的电子结构与几何结构不仅促进了水分子的解离生成H*,还可以降低HMF加氢的反应能垒,使其具有优异的反应活性与选择性。
该工作采用了简单的浸渍策略将1nm左右的磷钼酸团簇负载在自支撑的Cu纳米线上。通过球差校正高角度环形暗场扫描透射电镜对PMo12/Cu进行了微观结构的表征。Mapping表征结果图中均匀分布的Mo元素佐证了PMo12在Cu纳米线表面均匀分散。
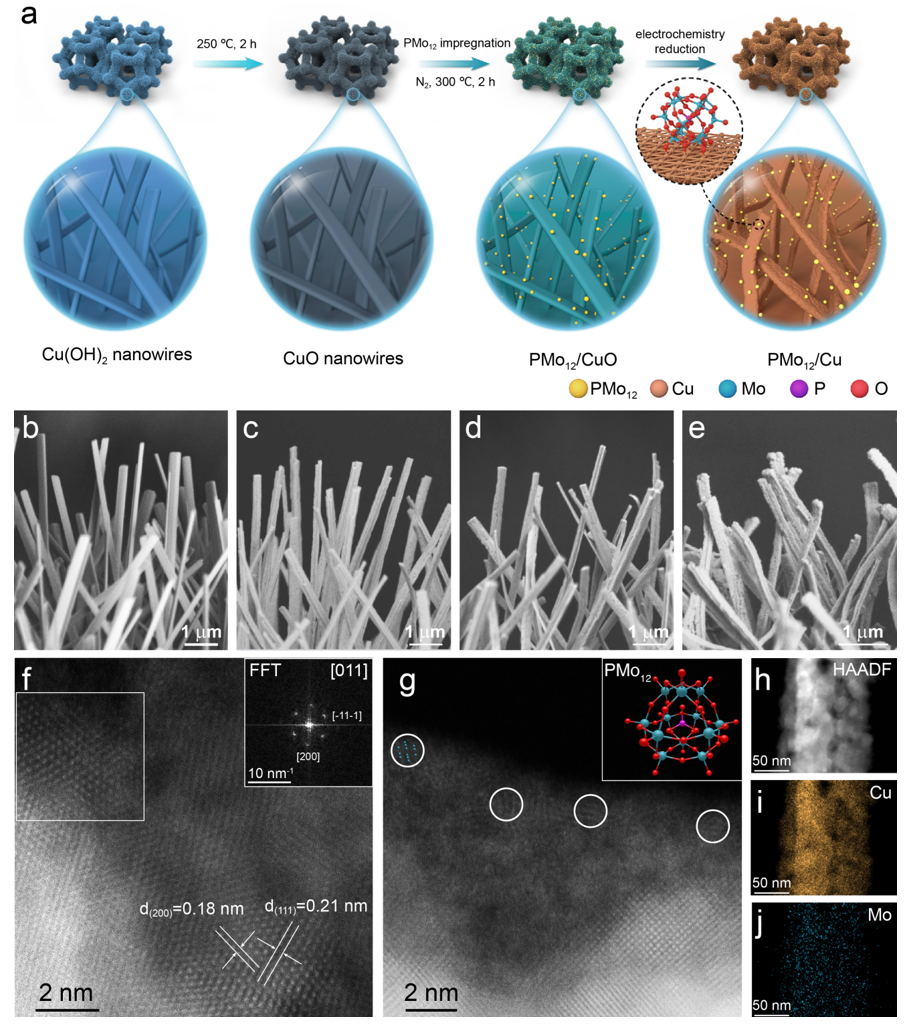
图1. (a) PMo12/Cu纳米线合成示意图以及产物的电镜表征图
研究进一步通过傅里叶变换红外光谱、拉曼光谱和X射线吸收精细结构谱表征发现负载在Cu纳米线上的PMo12团簇结构保持完整,并通过形成Mo−O−Cu键锚定在金属Cu表面。X射线光电子能谱进一步分析发现,PMo12/Cu表面存在更多的Cu+,且PMo12/Cu中的Mo元素的价态较PMo12/CuO中的Mo降低。PMo12/Cu的差分电荷密度分析进一步证实了形成异质结构后,Cu的电子会向PMo12团簇转移,导致异质界面处正电荷的积累。
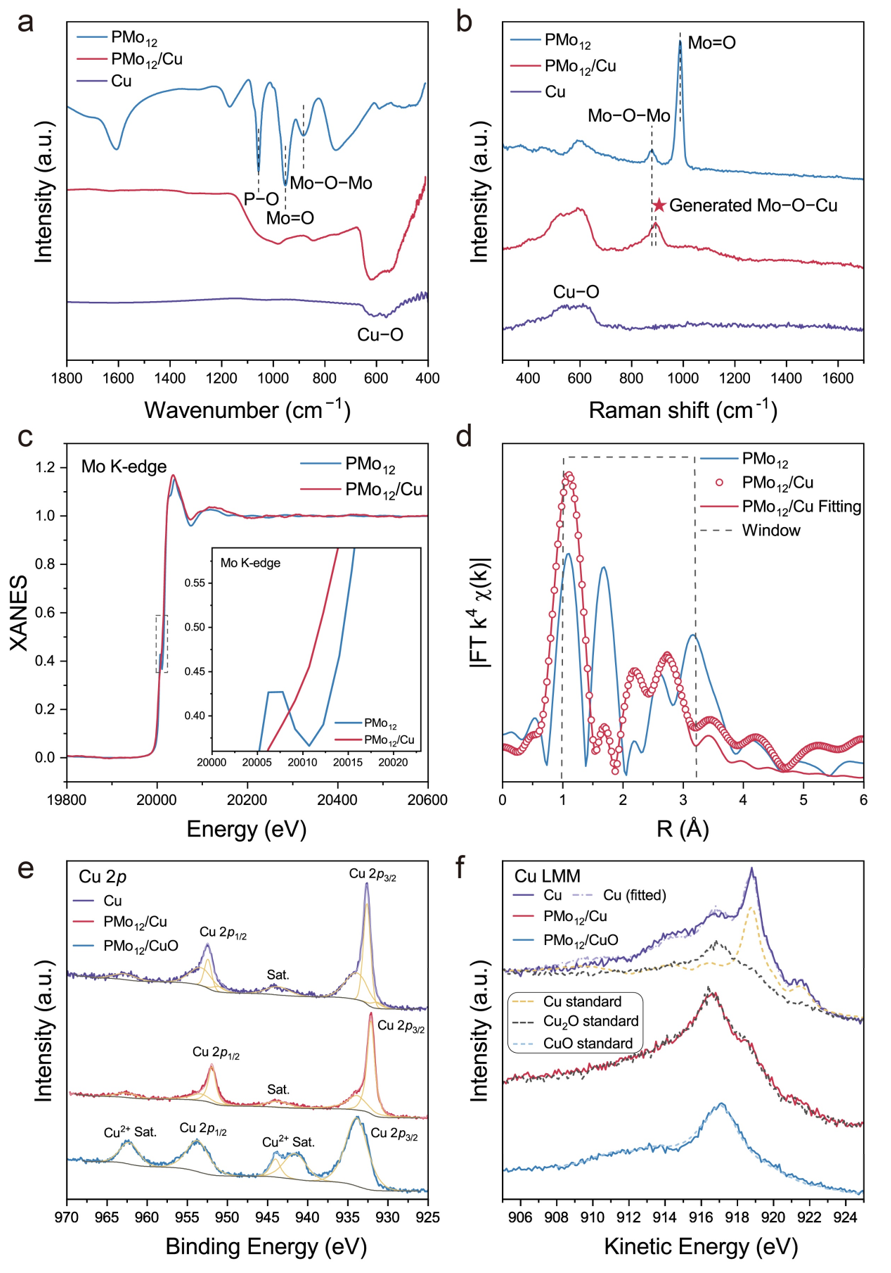
图2. PMo12、PMo12/Cu和Cu的红外、拉曼、XAS和XPS表征结果
团队将PMo12/Cu和Cu用于HMF的电化学加氢合成BHMF。由于HMF有多种可能的加氢反应方式,最终产物可能很复杂。PMo12团簇与Cu的结合后显著降低了HER和电化学加氢的过电位。在0.1 M HMF浓度下,通过计时电流法测试了催化剂加氢活性。对于Cu,在电位超过–0.25 V时生成耦合副产物,而在更负的电位下产生氢解产物。相反,PMo12/Cu显示出对目标产物BHMF的优异选择性,可以在较宽的电位范围内有效地获得BHMF。PMo12/Cu表现出对高浓度HMF显著的耐受性,即使在1.0 M的HMF浓度下,仍能保持99%的BHMF选择性。在如此高的浓度下,PMo12/Cu显示出高达98%的FE和4.45 mmol cm–2 h–1的BHMF生成速率。
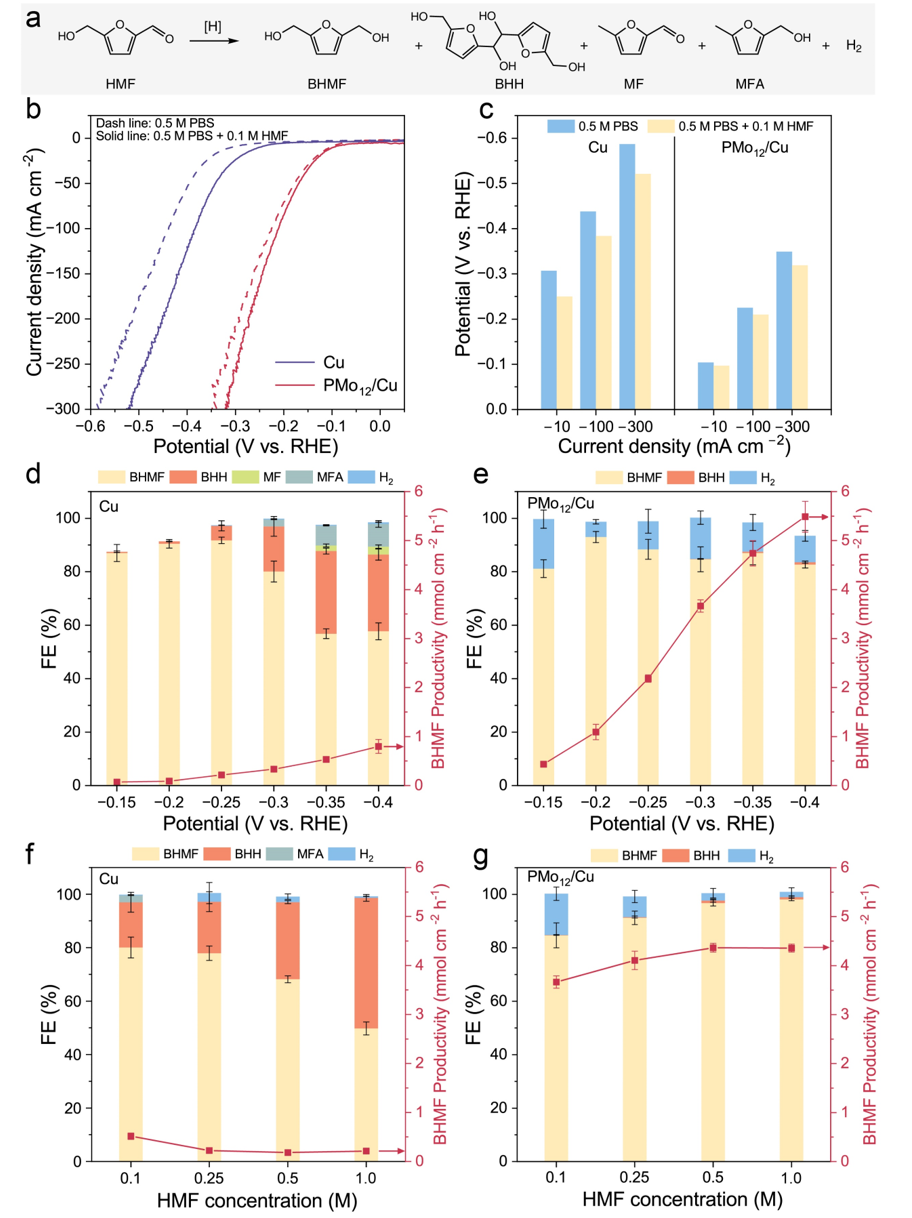
图3. Cu和PMo12/Cu的HMF电化学加氢性能对比
为进一步研究负载PMo12团簇对催化剂活性增强的原理,研究团队通过循环伏安测试以及准原位EPR测试发现HMF加氢涉及表面H*,且PMo12/Cu表面H*覆盖度更高。此外准原位EPR中发现在0.1 M HMF溶液中,Cu表面会产生了烷基自由基中间体,而PMo12/Cu并未观察到。表面增强的原位拉曼光谱结果表明,Cu催化剂在Volmer step后表面形成了吸附的OH–。而在PMo12/Cu上,并未发现相关振动峰,但出现了与Mo配位的氧与H+相互作用形成了OH或OH2基团的振动峰,表明在HER反应中,Volmer step更有可能发生在PMo12与Cu的界面处,并提供局部酸性环境,从而减少生成的OH–的吸附。在HMF电化学加氢的原位拉曼测试中发现PMo12在工作电位下增强了HMF的吸附。
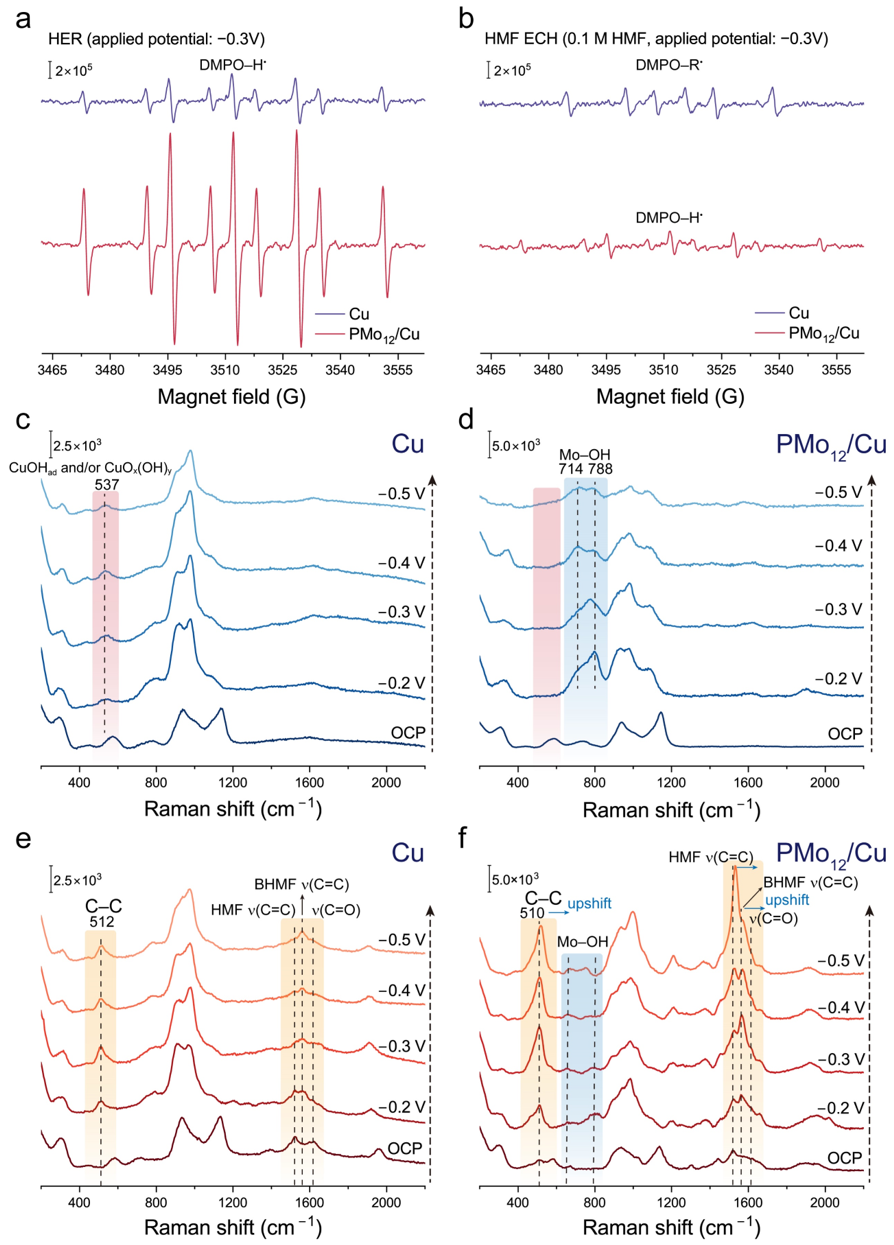
图4. Cu和 PMo12/Cu 的HER和HMF 电化学加氢的准原位EPR和原位拉曼光谱
随后团队深入研究了反应动力学。首先通过测试Tafel斜率发现,Cu和PMo12/Cu的HER动力学速控步是Volmer step。加入HMF之后Tafel斜率会降低,表明电化学加氢过程比HER过程更有利。氘代电解液中的EIS研究分析了电极界面处水分子的动力学行为。结果表明在PMo12与Cu形成异质界面后,H2O或D2O分子在催化剂表面的吸附和转移所需的驱动力更低。此外,氘代动力学同位素效应(KIE)实验结果表明HER和电化学加氢的RDS都涉及水分子中的H−OH键断裂,PMo12/Cu在HER中的KIE值比Cu小,这表明它对于H–OH键断裂的效率更高。
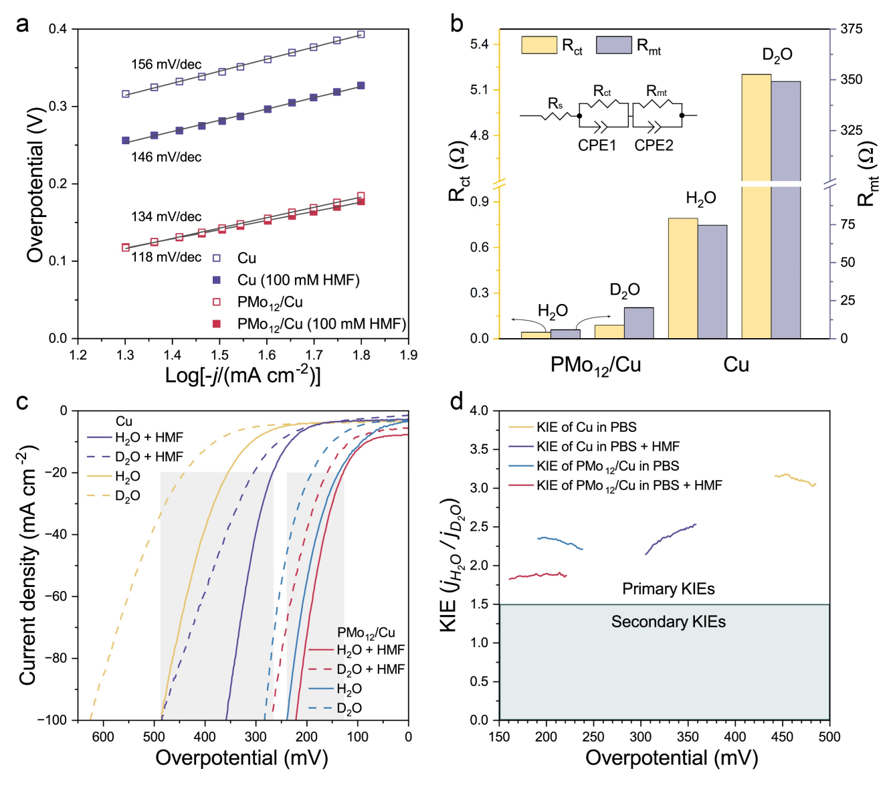
图5. Cu和PMo12/Cu的动力学研究
研究者通过DFT计算进一步研究了PMo12/Cu异质界面结构与其催化HER和HMF电化学加氢的构效关系。在PMo12/Cu催化剂上,H2O分子很容易发生H–OH键断裂。HMF加氢的反应中,发现两个催化剂中第一步加氢过程是反应速控步。然而,在第二步加氢中,PMo12/Cu的反应势垒比Cu低得多(0.25 eV 对比0.79 eV),这使得烷基自由基的加氢反应具有更快的动力学。相反,Cu表面会积累烷基自由基,从而引发偶联反应。分析第二步加氢过渡态结构发现,烷基自由基吸附在PMo12和Cu之间,并通过PMo12晶格氧的氢键而得到稳定。这种吸附结构以及与H*更短的距离是导致在烷基加氢反应中降低反应势垒的原因。
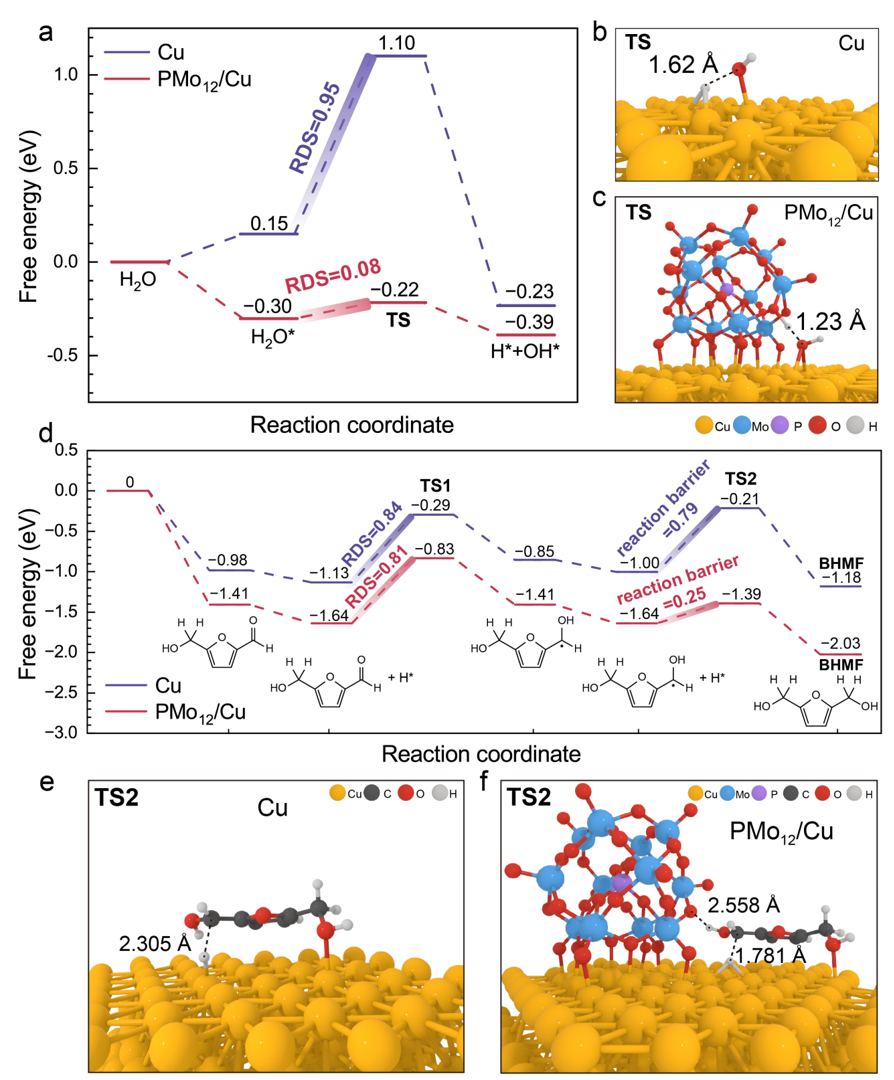
图6. PMo12/Cu和Cu上进行HER和HMF加氢的吉布斯自由能图,以及过渡态的结构图
总而言之,这个工作发展的PMo12/Cu的团簇级异质界面结构提供了独特的电子构型与几何结构,为增强水分子的吸附和活化以促进H*的生成,并为HMF第二步加氢反应提供更加有利的几何构型,显著降低了反应势垒。相关工作不仅为目前HMF电化学加氢选择性调控提供了新的策略,还为电催化剂异质界面工程调控催化活性与选择性带来了更深的认识。
上述研究得到国家重点研发计划、国家自然科学基金、西湖大学未来产业研究中心等的经费支持。
最新资讯














